Rétinoblastome
Kassimi FZ ; Labyad.S ; Sekhsoukh.R
Université Mohammed Premier d’Oujda, Faculté de médecine et de pharmacie d’Oujda ; CHU Mohammed VI d’Oujda, Maroc ; Laboratoire d’Oto-Neuro-Ophtalmologie LRONO ; Reçu le 03/10/2025 ; accepté le 04/11/2025 ; Disponible sur internet le 05/11/2025 ; DOI
Sommaire
Généralités :
Introduction
Définition
Intérêt
Épidémiologie
Physiopathologie et génétique
Diagnostic
Diagnostic positif
Examens complémentaires
Formes cliniques
Diagnostic différentiel
Évolution
Prise en charge thérapeutique
But
Moyens
Indication
Surveillance
Surveillance ophtalmologique
Surveillance pédiatrique
Pronostic
Médecine sociale
Impact psycho-socio-professionnel
Aspect médico-légal
Prévention et programme de santé
Conclusion
Annexe
Arbre décisionnel
Points importants/ à retenir
Généralités :
- INTRODUCTION :
- Il s’agit d’une tumeur rétinienne hautement maligne touchant le nourrisson et le jeune enfant.
- Elle est déterminée génétiquement et on lui décrit 2 formes : forme endophytique et forme exophytique .
- Les 2 principaux symptômes révélateurs : Leucocorie et le strabisme.
- Le Diagnostic est clinique par un FO.
- Le rétinoblastome est une tumeur guérissable si le dépistage et la PEC sont précoces.
- Intérêt : le pronostic vital peut être mis en jeu par l’extension orbitaire et les métastases d’où l’intérêt du dépistage précoce.
- EPIDEMIOLOGIE:
- Tumeur rare mais représente la tumeur intraoculaire primitive maligne la plus fréquente de l’enfant < 5ans.
- Elle peut être unilatérale (60 %) ou bilatéral (40 %).
- L’âge moyen de diagnostic varie dans les formes bilatérales, souvent avant 1 an, dans les formes unilatérales plus tardif (2 ans) selon les séries .
- Le gène du rétinoblastome siège sur le chromosome 13 q1-4 .
- 2 Types de rétinoblastome : Forme sporadique la plus fréquente (60%) et forme héréditaire : 40 %.
- PHYSIOPATHOLOGIE ET GENETIQUE :
Le gène suppresseur de tumeur dans lequel se produisent des mutations prédisposant au rétinoblastome est RB1 ; plus de 900 mutations différentes ont été identifiées à ce jour. La taille de la délétion génétique a tendance à être corrélée avec l’agressivité du rétinoblastome.
1-Le gène RB1 :
- Localisation : chromosome 13q14.
- Rôle physiologique : le gène RB1 code pour la protéine du rétinoblastome (pRB), un essentiel au contrôle du cycle cellulaire.
2-Fonction de pRB :
- En situation normale, pRB se lie au facteur de transcription E2F, l’empêchant d’activer les gènes nécessaires à la phase S.
- Quand la cellule reçoit un signal de prolifération, pRB est phosphorylée, libérant E2F → la cellule passe en phase S → réplication de l’ADN.
Ainsi, pRB bloque la division cellulaire tant que la cellule n’est pas prête à se diviser.
3-Mécanisme de cancérogenèse : l’hypothèse des deux coups (Knudson, 1971)
Pour qu’un rétinoblastome se développe, les deux allèles du gène RB1 doivent être inactivés.
- Premier « coup » : perte ou mutation d’un allèle germinale héritée ou de novo (constitutionnelle ou somatique).
- Deuxième « coup » : perte ou mutation du second allèle dans une cellule rétinienne.
4-Types de Retinoblastome :
a. Rétinoblastome héréditaire :
- Type de mutation : mutation germinale (constitutionnelle) du gène RB1, présente dans toutes les cellules de l’organisme.
- Transmission : autosomique dominante, avec pénétrance incomplète (~90 %).
- Atteinte oculaire : souvent bilatérale et multifocale (plusieurs foyers tumoraux dans chaque œil).
- Âge de survenue : plus précoce (généralement avant 1 an).
- Mécanisme génétique : Premier ou Deuxième « coup » .
- Risques associés :
- Risque de tumeurs secondaires (ostéosarcome, sarcome des tissus mous, mélanome, etc.).
- Risque de transmission à la descendance (50 %).
- Autres particularités : possibilité d’un rétinoblastome trilatéral (atteinte pinéale associée) .
b. Rétinoblastome sporadique :
- Type de mutation : deux mutations somatiques acquises du gène RB1 survenant uniquement dans une cellule rétinienne.
- Transmission : non héréditaire, non transmissible à la descendance.
- Atteinte oculaire : le plus souvent unilatérale et unifocale.
- Âge de survenue : plus tardif (souvent entre 2 et 5 ans).
- Mécanisme génétique :
- Premier et deuxième « coups » : mutations somatiques indépendantes sur les deux allèles de RB1 dans une même cellule rétinienne.
- Risques associés :
- Pas de risque accru de second cancer.
- Pas de risque pour la fratrie ou les descendants.
- Autres particularités : forme la plus fréquente (≈ 60 % des cas).
Diagnostic :
A. DIAGNOSTIC POSITIF :
- Signes d’appel :
- Les 2 signes révélateurs majeurs :
- La leucocorie : reflet pupillaire blanc ; inconstante au début de la maladie(Constatée par les parents ou parfois découverte fortuite lors d’un cliché photo où l’on constate un reflet blanc).
- Le strabisme : strabisme unilateral permanent, signe d’appel précoce et témoigne d’une atteinte maculaire.
- Autres signes plus rares : baisse de l’acuité visuel avec Nystagmus, buphtalmie, exophtalmie, cellulite orbitaire,uvéite, hypopion , rubéose irienne, hyphéma, association avec une cataracte congénitale, persistance du vitré primitif, microphtalmie .
- Dépistage systématique : Si anomalies impliquant le chromosome 13 ou si antécédents familiaux de RTB.
- Interrogatoire : précise :
– L’âge de l’enfant, date d’apparition et évolution des signes.
– ATCD familiaux de RTB.
- Examen ophtalmologique :
- Le test des reflets cornéens de Hirschberg : à l’aide d’un ophtalmoscope direct, est un test de dépistage simple de la leucocorie.
- Le diagnostic repose essentiellement sur l’examen du FO sous anesthésie général avec dilatation qui met en évidence : Le rétinoblastome endophytique apparaît comme une tumeur blanche richement vascularisée se développant vers la cavité vitréenne avec de nombreux flocons blanchâtres flottant dans le vitré. Les formes exophytiques se présentent sous forme d’un décollement de rétine derrière lequel on aperçoit les masses saillantes parfois calcifiées en partie blanches avec dilatation angiomateuse des vaisseaux.
- Le bilan précis des lésions oculaires permet de classer le rétinoblastome selon l’International Retinoblastoma Classification :
Groupe A : bon pronostic tumoral et visuel.
- Tumeurs < 3 mm.
- Situées à >3 mm de la fovéola et >1,5 mm de la papille.
- Pas d’essaimage dans le vitré ou en sous-rétinien.
Groupe B :
- Tumeurs ≥ 3 mm.
- Décollement séreux inférieur < 5 mm autour de la tumeur.
- Pas d’essaimage dans le vitré ou en sous-rétinien.
Groupe C :
- Essaimage vitréen ou sous-rétinien localisé.
- DSR ≥ 5 mm de la base de la tumeur jusqu’à un quadrant.
Groupe D :
- Essaimage vitréen massif (boules de neige) et/ou sous-rétinien diffus et massif.
- Décollement de rétine > 1 quadrant.
Groupe E : pas de conservation oculaire possible.
Globe sans potentiel visuel avec présence d’un ou plusieurs signes suivants :
- Hémorragie intra-vitréenne massive.
- Glaucome néo-vasculaire
- Rétinoblastome infiltrant diffus.
- Tumeur en avant de la hyaloïde antérieure.
- Tumeur touchant le cristallin.
- Phtyse du globe.
- Cellulite orbitaire aseptique.
- Examen général : par un pédiatre (abdominal, neurologique, aires GG, ostéoarticulaire …).
- Conseil génétique : Histoire familiale, avec examen des membres de la famille et arbre généalogique.
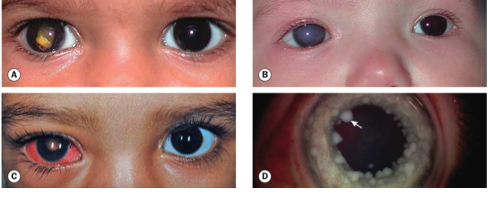
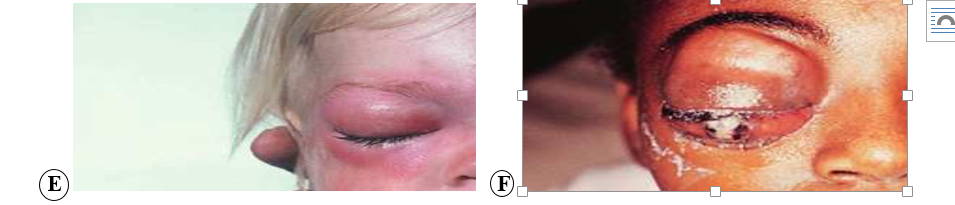
Fig.1 : Modes de découverte du rétinoblastome. (A) Leucocorie ; (B) glaucome secondaire et buphtalmie ; (C) yeux rouges dus à une uvéite ; (D) nodules iriens (flèche) ; (E) inflammation orbitaire ; (F) invasion orbitaire.
- EXAMENS COMPLEMENTAIRES :
1-Échographie mode B :
Confirme le diagnostic si forme atypique met en évidence une masse hyperéchogène intra-vitréenne contenant de fines calcifications dans 95 % des cas.
2-Bilan d’extension:
2.1-L’IRM orbito-cérébrale :
– Meilleur bilan d’extension objective l’extension vers :
- Le nerf optique ;
- La choroïde ;
- La paroi orbitaire et sclérale ;
- L’extension intracrânienne.
- Le RTB est Hyperintense T 1 , Hypointense T2, se rehaussant après injection du gadolinium .
- Recherche l’association rare avec un Pinéaloblastome (tumeur trilatérale).
2.2-Anatomopathogie de la pièce d’énucléation :
Confirme le diagnostic, précise la différentiation et l’extension vers les tuniques de l’œil, et recherche des facteurs histopronostiques.
- Aspect macroscopique :
- Masse blanchâtre, friable, intrarétinienne.
- Zones de nécrose et de calcifications fréquentes.
- Histologie :
- Petites cellules rondes indifférenciées (aspect « en ciel étoilé »).
- Présence de rosettes de Flexner-Wintersteiner (typique).
- Parfois rosettes d’Homer Wright (moins spécifiques).
2.2-Bilan d’extension à distance :
Si tumeur extra oculaire ou intra oculaires avec Facteurs histopronostic de métastases élevées ou moyens : IRM cérébral , ponction lombaire, myélogramme et biopsie médullaire , échographie abdominale ; + /- Rx thorax.
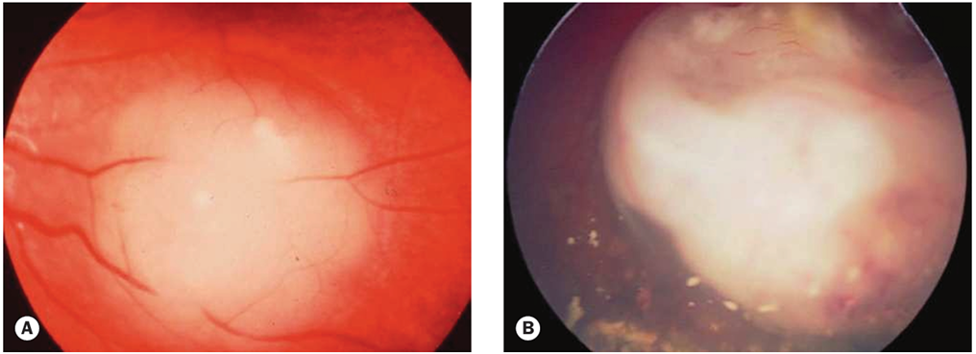
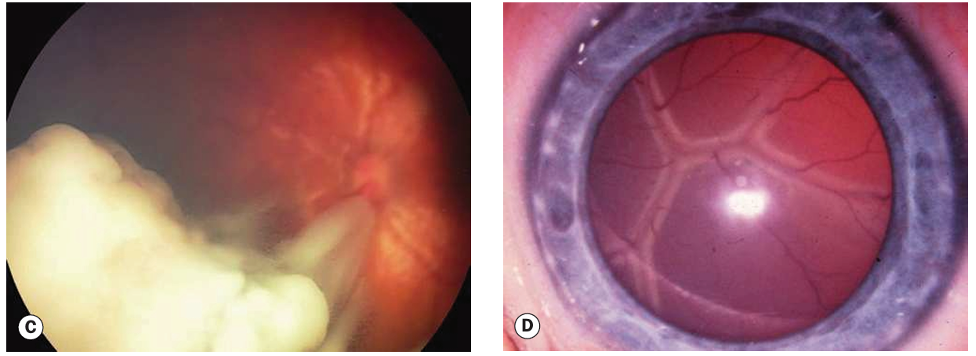
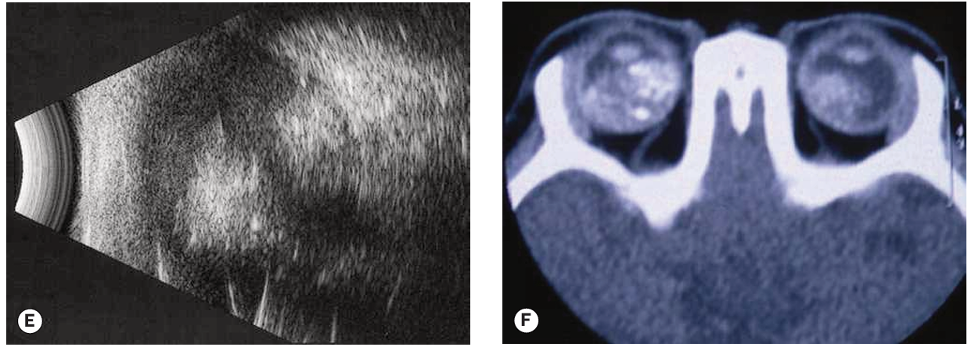
Fig.2 : Rétinoblastome. (A) Tumeur intrarétinienne ; (B) tumeur endophytique avec ensemencement vitréen ; (C) croissance endophytique et exophytique mixte ; (D) décollement total de la rétine ; (E) B-scan montrant les calcifications hyperéchogènes ; (F) TDM axiale montrant des tumeurs bilatérales avec calcification.
- FORMES CLINIQUES:
3.1-Formes anatomo-cliniques :
- RTB endophytique : La plus fréquente, envahissement progressif du vitré.
- RTB exophytique : Développement sous rétinien avec envahissement de la choroïde et la sclère , décollement rétinien exsudatif localisé autour de la tumeur ou total.
- RTB infiltrant diffus : très rare, il n’y a pas de masse tumorale rétinienne classique. Les enfants présentent souvent une atteinte du segment antérieur avec aspect de pseudo-uvéite, parfois des nodules iriens, une néo-vascularisation de l’iris y est parfois associée. Au FO, il peut exister des éléments nodulaires dans le vitré qui peuvent faire évoquer une pathologie inflammatoire. La rétine est souvent infiltrée et blanchâtre, épaissie en échographie ou imagerie oculaire par IRM, mais sans masse tumorale rétinienne classique.
3.2-Formes associées : à d’autre Tumeur maligne (pinéaloblastome, lymphome) ou anomalies chromosomiques (trisomie 13 ou 21 ou Délétion du chromosome13,) .
D. DIAGNOSTIC DIFFERENTIEL :
1-Maladie de Coats évoluée :
Angiomatose retinienne idiopathique , caractérisée par des télangiectasies rétiniennes responsables de l’apparition des exsudats jaunâtres intra et sous-rétiniens parfois peseudotumoraux et dont l’évolution spontanée se fait vers un décollement rétinien (DR) total.
2-Toxocarose :
-Uvéite postérieure d’origine parasitaire caractérisée par des travées et des granulomes rétiniens profonds .
-Survenue dans un contexte de contact avec les chiens
-Diagnostique serologique .
3-Autres Tumeurs rares :
–Astrocytome rétinien : souvent associée à la sclérose tubéreuse de Bourneville.
–Médulloépithéliome : tumeur primitive de l’épithélium ciliaire, rare.
–Retinome (rétinocytome) est une variante du rétinoblastome qui est généralement bénin mais avec un
profil génétique indiquant une lésion précancéreuse – rarement, un rétinome peut subir une transformation
tardive en un rétinoblastome à crois sance rapide. Il se manifeste par une lésion lisse et blanchâtre, en forme
de dôme, qui évolue généralement spontanément en une masse calcifiée associée à une altération de l’EPR et
une atrophie choriorétinienne .
4-Autres causes non tumorales :
– La dysplasie vitréorétinienne est causée par une différenciation défectueuse de la rétine et du vitré qui se traduit par une rétine dysplasique détachée, formant une masse rétrolentale avec leucocorie.
-Persistance du vitré primitif
-DR non tumoral
-Rétinopathie du prématuré (ROP)
-Colobomes.
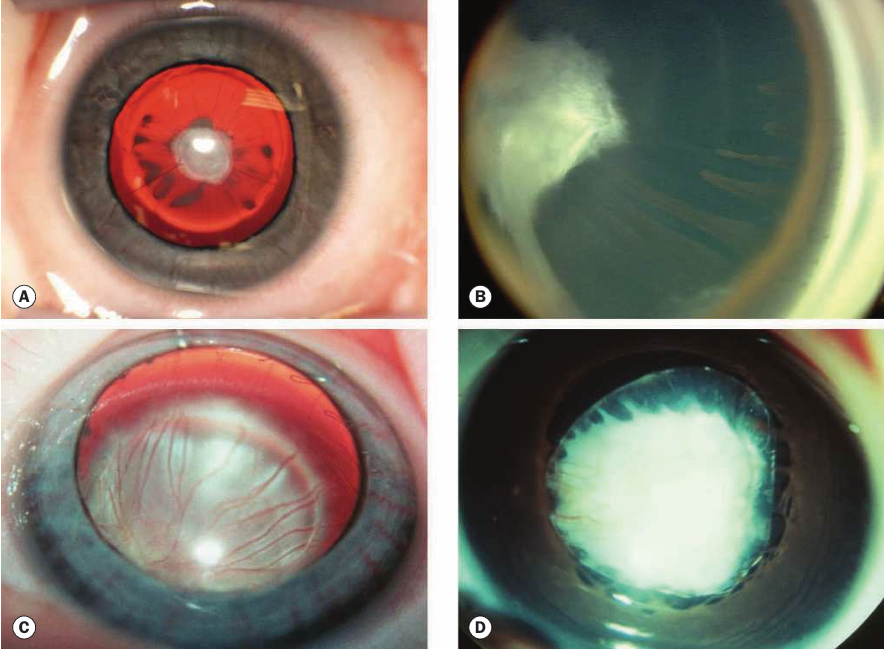
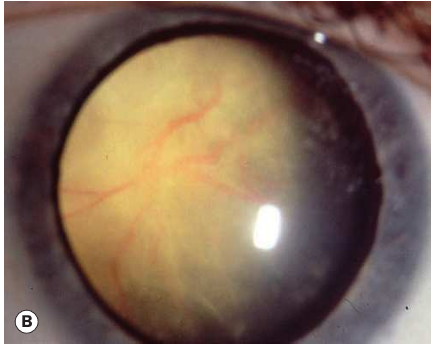
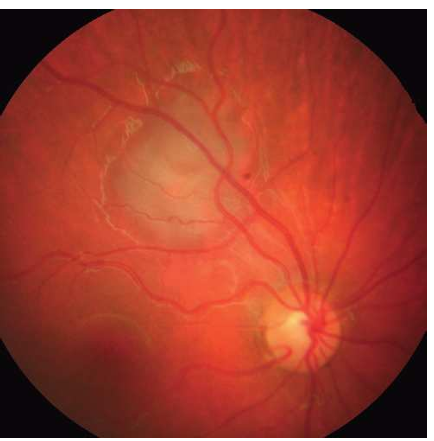
Fig.2 : Diagnostics différentiels du rétinoblastome : Vascularisation fœtale antérieure persistante (A,B,C et D) : (A) Cataracte polaire antérieure avec vasculari sation radiaire ; (B) masse rétrolentale avec processus ciliaires allongés ; (C) vaisseaux rétrolentaux et fibrose moins dense ; (D) plaque dense avec modifications secondaires du cristallin. (E) Dysplasie vitréorétinienne; (F) Rétinome.
- EVOTUTION :
1-Spontanée :
– Extension orbitaire et métastatique.
– Involution spontanée rare (à rechercher chez les parents : cicatrices au FO).
2-Sous traitement :
– Guérison .
– Récidives oculaire ou orbitaire après énucléation, bilatéralisation secondaire.
Prise en charge thérapeutique :
- La prise en charge doit être pluridisciplinaire : ophtalmologue pédiatrique, oncologue pédiatrique, radiothérapeute, généticien, anesthésiste.
- Le traitement doit être individualisé, en fonction de nombreux facteurs : âge, bilan des deux yeux, statut génétique RB1, potentiel visuel, extension, ressources disponibles
A. But :
- Préserver le pronostic vital en évitant la dissémination systemique et intra-cranienne de la tumeur .
- Préserver le pronostic fonctionnel : éviter l’énucléation si un traitement conservateur est possible et sûr.
- Conseil génétique : Assurer le suivi génétique et prévenir les tumeurs malignes secondaires notamment dans les formes héréditaires liées à une mutation du gène RB1.
B. Moyens :
- TRT chirurgical :
- Énucléation : Doit être réalisée avec un minimum de manipulations et il est impératif de retirer une section du nerf optique d’au moins 10 mm.
- Exentération orbitaire .
- TRT médical :
- Chimiothérapie :
C’est le pilier du traitement dans la plupart des cas et peut être utilisée en conjonction avec des traitements locaux :
- Systémique : le carboplatine, l’étoposide et la vincristine (CEV) sont administrés en trois à six cycles selon le stade du rétinoblastome. La chimiothérapie avec une seule molécule (carboplatine seule) ou deux a également donné des résultats favorables dans certaines circonstances, en tant que traitement transitoire en attendant un traitement plus agressif.
- Chimiothérapie sélective intra-artérielle de l’artère ophtalmique : il s’agit d’une nouvelle option thérapeutique prometteuse, avec des taux élevés de préservation du globe oculaire.. Dans cette procédure, une canule de petit calibre est introduite via l’artère fémorale jusqu’à l’artère ophtalmique. Le traitement offre des résultats nettement meilleurs que le trai tement intraveineux dans les yeux classés dans le groupe D. Le melphalan ou le topotécan sont ensuite injectés pendant environ 30 minutes vers les artères qui vascularisent l’uvée et la rétine. Le profil de sécurité global est meilleur que la chimiothérapie intraveineuse ;
- Le melphalan intravitréen semble être efficace en cas d’ensemencement vitréen, bien qu’il existe un risque faible de dissémination extra-oculaire ;
- la chimio-réduction peut être suivie d’un traitement focal par cryothérapie ou thermothérapie transpupillaire.
- Cryoapplication :
Cryoapplication de la tumeur sous contrôle ophtalmoscopique : utilisant une technique de triple cycle de congélation–décongélation 2 à 3 séances à 1 mois d’intervalle.
- Thermothérapie transpupillaire (TTT):
- Irradiation de chaque site tumorale par un laser diode hyperthérmie cytotoxique.
- Utilisée avec la chimiothérapie ou parfois employée de façon isolée.
- Radiothérapie :
- Curiethérapie :
Irradier la tumeur par une plaque radioactive suturée à la sclère .
- Radiotherapie externe :
Elle doit être évitée si possible, en particulier chez les patients atteints de rétinoblastome héréditaire, en raison du risque de seconde tumeur maligne en particulier les sarcomes ainsi que d’autres complications : retentissement sur la croissance orbito-faciale, l’ostéonécrose, la cataracte, la rétinopathie radique, la neuropathie radique, insuffisances hypophysaires .
C. Indications :
- Traitement conservateur :
Indiqué chez tous les RTB bilat et chez 20 % des RTB unilateraux .
- Chimiothérapie néo-adjuvante : SIle volume tumoral initial ne permet pas l’accessibilité au TRT conservateurs.
- Tumeurs en avant de l’équateur de l’œil :
- Cryoapplication : Petite tumeurs (< 3 mm), sans envahissement profond ni ensemencement du vitré:
- Curiethérapie :Tumeurs > 3mm en l’absence d’ensemencement vitréen et dans d’autres circonstances telles que la résistance à la chimiothérapie .
- Tumeurs en arrière de l’équateur de l’œil :
- Chimio-thérmo thérapie ou TTT seule.
- Tumeurs étendues de la rétine ou du vitré :
- Chimiothérapie par cathétérisme de l’artère ophtalmique : Diminue le recours à l’énucléation et/ou à l’irradiation externe.
- Radiothérapie externe : en cas de rétinoblastome résiduel ou récidivant après une chimiothérapie systémique ou intra-artérielle dans l’artère ophtalmique.
- Traitement non conservateur : si RTB très étendu.
- Chimiothérapie néo-adjuvante : cas très avancés avec buphtalmie(limiter les risques de rupture oculaire peropératoire) et/ou atteinte du NO détectable en imagerie (afin de permettre une section du NO en zone saine.
- L’énucléation sous AG avec mise en place d’implant : est généralement indiquée en cas de glaucome néovasculaire, d’envahissement de la chambre antérieure et/ou du nerf optique ou si la tumeur occupe plus de la moitié du volume vitréen (groupe E). Elle est également envisagée en cas d’échec de la chimio-réduction et est utile pour le rétinoblastome diffus en raison d’un mauvais pronostic visuel et d’un risque élevé de récidive avec d’autres modalités thérapeutiques
- L’analyse histopathologique de la pièce d’énucléation permet de poser l’indication du Traitement adjuvant :
- 4 cures de Chimiothérapie adjuvante pour les groupes à risque moyen .
- 6 cures Chimiothérapie adjuvante + radiothérapie orbitaire pour les groupes à haut risque.
- Récidive orbitaire après énucléation : radiothérapie orbitaire + chimiothérapie +/- exentération.
- Récidive oculaire après traitement conservateur : Cryoapplication ou curithérapie. Si échec, énucléation.
- Métastases : énucléation + chimiothérapie + radiothérapie orbitaire.
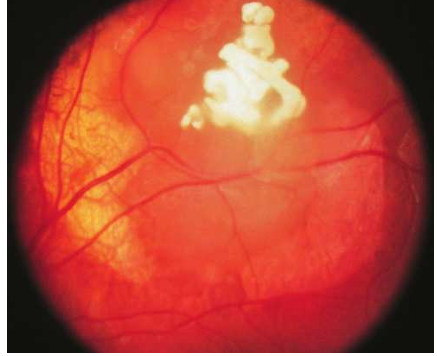
Fig .4 : Régression de la tumeur après curithérapie .
Suveillance :
- Surveillance ophtalmologique :
- FO : 1×/ mois puis 1× / 3mois l’adolescence 1×/6mois ; puis annuel à vie.
- Rechercher :
- une récidive tumorale,
- une atteinte controlatérale,
- Toxicité du TRT local (cataracte, rétinopathgie radique… )
- le suivi de l’appareillage.
- Surveillance pédiatrique :
- PEC de l’handicap visuel
- Recherche de toxicité du TRT et d’une Tumeur IIairechez les enfants porteurs d’une anomalie du gène RB1.
C. Pronostic :
- Pronostic vital : dépend de :
- Âge : plus l’enfant est jeune plus il est mauvais.
- Délai entre début d’évolution et le traitement.
- D egré d’extension.
- Bilatéralité.
- Degré de différenciation : indifférencié ® mauvais pronostic.
- Formes héréditaires de RTB : risque de survenue de Tm secondaire.
- Pronostic fonctionnel :
- Dépend du volume initial de la tumeur, sa distance par rapport à la macula et du résultat du traitement conservateur.
Médecine sociale:
A–Impact psycho-socio-professionnel :
a.Impact psychologique :
- Diagnostic de cancer chez un nourrisson : choc émotionnel majeur.
- Anxiété parentale, culpabilité, peur de la cécité ou de la récidive.
- Altération de l’image corporelle après énucléation (prothèse oculaire).
- Importance d’un accompagnement psychologique familial.
b.Impact social :
- Hospitalisations fréquentes → désorganisation familiale.
- Difficultés d’accès à l’école pour l’enfant atteint d’une déficience visuelle.
- Besoin de rééducation visuelle et de soutien scolaire.
c.Impact professionnel (parents) :
- Arrêts de travail répétés.
- Contraintes économiques (déplacements, soins spécialisés).
- Importance du soutien social et de l’aide administrative (ALD, MDPH)
B-Aspect médico-légal :
- Déclaration obligatoire en cas de maladie génétique familiale.
- Conseil génétique à proposer à chaque famille :
- Étude moléculaire du gène RB1.
- Dépistage des apparentés à risque (fond d’œil néonatal).
- Consentement éclairé indispensable pour :
- Les prélèvements génétiques.
- Les traitements lourds (chimiothérapie, énucléation).
- Obligation d’informer les parents sur le risque héréditaire et les possibilités de diagnostic prénatal (ou préimplantatoire).
C-Prévention et programmes de santé :
a.Prévention primaire :
- Pas de prévention spécifique du rétinoblastome sporadique.
- Dépistage génétique possible dans les familles à mutation RB1 connue.
- Diagnostic préimplantatoire envisageable (en FIV) pour éviter la transmission.
b. Prévention secondaire:
- Dépistage précoce :
- Fond d’œil systématique chez les enfants à risque (fratrie).
- Sensibilisation des parents à la leucocorie sur photo (campagnes d’information).
- Formation des pédiatres et médecins de famille au réflexe pupillaire anormal.
- Le risque de transmission à la descendance est de 50 % dans les formes familiales, et de 5 % dans les formes sporadiques.
- La M.E.E d’une mutation constitutionnelle du gène RB1 chez un patient tests génétiques chez ses apparentés.
c. Prévention tertiaire:
- Surveillance à vie pour prévenir les seconds cancers.
- Éviter les irradiations inutiles chez les porteurs d’une mutation RB1.
- Rééducation visuelle et accompagnement social.
Conclusion :
- Le rétinoblastome est une tumeur dont le pronostic vital reste sombre dans les pays en voie de développement faute de diagnostic précoce et d’une prise en charge adaptée.
- Dans les pays développés, son pronostic est excellent avec de gros progrès réalisés dans le domaine des traitements conservateurs.
- Le rétinoblastome nécessite une PEC multidisciplinaire ainsi qu’un suivi spécialisé à long terme.
Annexes :
A- Arbre décisionnel thérapeutique :
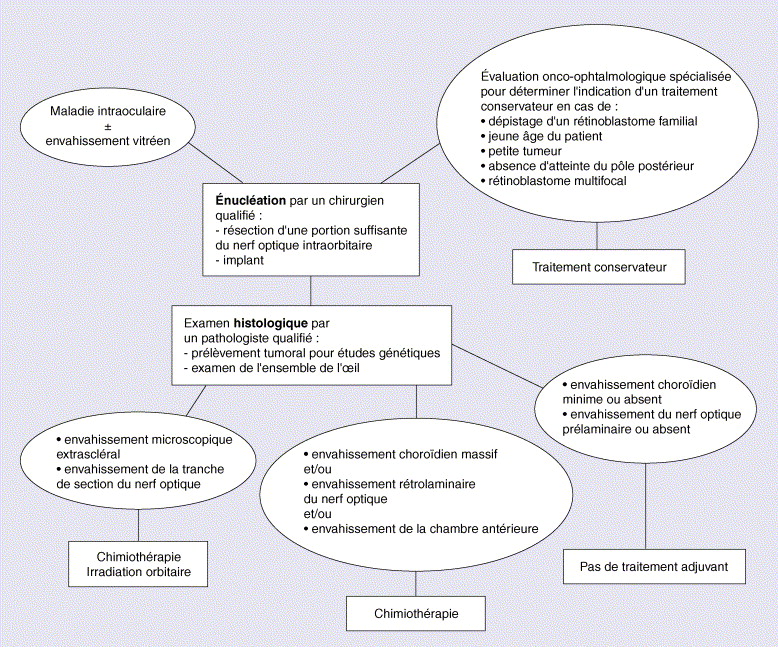
B- Points importants/ à retenir :
- Tumeur maligne intraoculaire la plus fréquente chez l’enfant, souvent avant 5 ans.
- Signes évocateurs : leucocorie (œil blanc) et strabisme.
- Diagnostic : examen du fond d’œil sous anesthésie + imagerie (échographie, IRM).
- Cause : mutation du gène RB1, héréditaire (bilatéral) ou sporadique (unilatéral).
- Importance du bilan génétique et du suivi régulier pour prévenir récidives et cancers secondaires.
- Traitement adapté au stade (classification ICRB) :
- Groupes A-B : traitements conservateurs locaux.
- Groupes C-D : chimiothérapie ± traitement local.
- Groupe E : énucléation.